Richtlinien zur US-Zertifizierung
FDA-Validierung der Cybersicherheit in Medizingeräten
Die FDA hat ihre Richtlinien zur Softwarevalidierung mit zusätzlichen Anforderungen an die Cybersicherheit ergänzt. Für MedTech-Hersteller ist die statische Analyse die effektivste Methode, um sich mit vorhersagbarer Software den Sicherheitsbedrohungen zu stellen und die Regulatorik zu erfüllen.
Im September 2023 hat die FDA einen neuen Leitfaden zur Cybersicherheit für Medizinprodukte veröffentlicht. Er enthält weitreichende Empfehlungen für das Design von Cybersicherheit sowie die Kennzeichnung und die Dokumentation, die für medizinische Produkte mit Cybersicherheitsrisiken vor dem Inverkehrbringen zu berücksichtigen sind.
Der Leitfaden soll Herstellern helfen, Cybersicherheitsrisiken während des gesamten Lebenszyklus eines Medizinprodukts zu identifizieren und zu reduzieren, von der Konzeption und Entwicklung über die Herstellung und Wartung bis hin zur Überwachung nach dem Inverkehrbringen. Zudem beinhaltet er Empfehlungen für die Kommunikation von Cybersicherheitsrisiken an Gesundheitsdienstleister und Patienten.
Der neue FDA-Leitfaden empfiehlt
Der neue Leitfaden baut auf früheren FDA-Leitfäden zur Cybersicherheit auf, ergänzt um eine Reihe neuer und aktualisierter Empfehlungen, wie beispielsweise folgende Anforderungen an die MedTech-Hersteller:
- Einführung eines umfassendes Programms zum Management von Cybersicherheitsrisiken
- Vorlage einer Software Bill of Materials (SBOM) für alle Medizingeräte
- Entwicklung und Umsetzung von Cybersicherheitsplänen für die Zeit nach der Markteinführung
- Empfehlungen für die Kommunikation von Cybersicherheitsrisiken an Gesundheitsdienstleister und Patienten
Während sich die FDA in der Vergangenheit auf Aspekte der funktionalen Sicherheit von Systemen konzentriert hat, ist Cybersecurity heute ein ebenso wichtiges Thema. Obwohl Safety und Security ähnlich sind – und man könnte leicht argumentieren, dass es bei beiden um die Erstellung vorhersehbarer Software geht – erfordert die Cybersecurity laut FDA besondere Aufmerksamkeit und Maßnahmen.
Prozessvalidierung für Medizinprodukte
Unter Prozessvalidierung versteht man die Sammlung und Auswertung von Daten, die den wissenschaftlichen Nachweis erbringen, dass ein Prozess in der Lage ist, ein qualitativ hochwertiges Produkt herzustellen. Im Zusammenhang mit Medizinprodukten ist die Prozessvalidierung von entscheidender Bedeutung, um die Sicherheit und Wirksamkeit der Produkte während ihres gesamten Lebenszyklus zu gewährleisten.
Sie ist besonders wichtig für softwaregesteuerte Medizinprodukte, da Software komplex und schwer umfassend zu testen sein kann. Beispielsweise kann ein softwaregesteuertes Medizinprodukt Millionen von Codezeilen umfassen, und es ist nicht möglich, alle möglichen Szenarien und Kombinationen von Eingaben und Ausgaben zu testen, ohne sowohl eine statische Codeanalyse als auch eine dynamische Analyse durchzuführen.
Es gibt drei Hauptgründe, warum Prozessvalidierung wichtig ist:
- Qualitätssicherung: Die Prozessvalidierung trägt dazu bei, dass Medizinprodukte konsistent und zuverlässig hergestellt werden, wodurch das Fehlerrisiko verringert und ein hohes Qualitätsniveau gewährleistet wird.
- Patientensicherheit: Durch die Validierung des Herstellungsprozesses können potenzielle Risiken und Fehler erkannt und reduziert werden, was die Patientensicherheit erhöht.
- Einhaltung gesetzlicher Vorschriften: Das Einhalten gesetzlicher Vorschriften, wie z. B. der FDA Quality System Regulation (QSR), erfordert den Einsatz validierter Prozesse. Ist dies nicht der Fall, kann es zu Compliance-Problemen kommen.
Gesetzliche Anforderungen
Die regulatorischen Anforderungen, die im FDA-Leitfaden zur Cybersicherheit von Medizinprodukten dargelegt sind, sollen den wachsenden Bedenken hinsichtlich der Sicherheit von Medizinprodukten in einer zunehmend vernetzten und digitalen Gesundheitsumgebung Rechnung tragen. Hier findet man eine Erläuterung der regulatorischen Anforderungen.
Gesetzliche Befugnisse
Die FDA hat im Rahmen des Food and Drug Omnibus Reform Act (FDORA), der im Dezember 2022 in Kraft getreten ist, neue gesetzliche Befugnisse erhalten. Dieses Gesetz ermöglicht es der FDA, bei der Einreichung von Medizinprodukten Informationen zur Cybersicherheit zu verlangen und verpflichtet die Hersteller, bestimmte Maßnahmen zu ergreifen, um nachzuweisen, dass ihre Produkte und die zugehörigen Systeme cybersicher sind. Die Nichteinhaltung dieser Anforderungen gilt als verbotene Handlung, die strafrechtlich verfolgt werden kann.
Definition »Cybergerät«
Im Gesetz wird der Begriff »Cybergerät« neu definiert als ein Gerät, das vom Hersteller validierte, installierte oder autorisierte Software enthält, mit dem Internet verbunden werden kann und technologische Merkmale aufweist, die für Bedrohungen der Cybersicherheit anfällig sein können. Diese Definition trägt zur Klärung der Frage bei, welche Geräte unter die neuen Cybersicherheitsanforderungen fallen.
Cybersicherheit in der Zulassung
Nach den neuen Richtlinien müssen Hersteller bestimmte Informationen zur Cybersicherheit in ihre Zulassungsanträge bei der FDA aufnehmen. Diese Premarket-Anträge können Premarket-Approval (PMA), 510(k)- oder De-Novo-Anträge sein. Die Informationen zur Cybersicherheit sind erforderlich, um sicherzustellen, dass das Produkt die gesetzlichen Cybersicherheitsanforderungen erfüllt.
Nach der Markteinführung
Produkthersteller sind verpflichtet, einen Plan zur Überwachung, Identifizierung und Behebung von Cybersicherheitsschwachstellen und Exploits nach dem Inverkehrbringen vorzulegen. Dieser sollte den Ansatz des Herstellers zur kontinuierlichen Überwachung und Behebung von Cybersicherheitsrisiken darlegen, die nach dem Inverkehrbringen des Produkts auftreten können.
Gewährleistung der Cybersicherheit
Hersteller müssen Prozesse und Verfahren konzipieren, entwickeln und aufrechterhalten, die hinreichende Gewähr dafür bieten, dass das Produkt und die zugehörigen Systeme cybersicher sind. Diese Anforderung unterstreicht die Notwendigkeit eines proaktiven Ansatzes bei der Integration von Sicherheit in das Produktdesign und deren Aufrechterhaltung während des gesamten Produktlebenszyklus.
Software Bill of Materials
Von den Herstellern wird erwartet, dass sie als Teil ihres Antrags eine Softwarestückliste (Software Bill of Materials, SBOM) vorlegen. Diese sollte die kommerziellen, quelloffenen und handelsüblichen Softwarekomponenten auflisten, die in dem Gerät zum Einsatz kommen. Diese Informationen sind entscheidend, um potenzielle Schwachstellen im Software-Stack zu identifizieren und zu beheben.
Zusatzanforderungen der FDA
Das Leitliniendokument ermächtigt die FDA auch, zusätzliche Anforderungen an die Cybersecurity von Produkten und zugehörigen Systemen durch Vorschriften festzulegen. Diese müssen von den Herstellern erfüllt werden.
Verbot bei Nichterfüllung
Ein wichtiger Aspekt der regulatorischen Anforderungen ist die Einführung eines neuen gesetzlichen Verbots: Die Nichteinhaltung der Cybersecurity-Anforderungen der FDA ist nicht nur eine Frage der Compliance, sondern stellt auch einen Gesetzesverstoß dar. Die Regierung ist befugt, Verstöße gegen diese Anforderungen strafrechtlich zu verfolgen oder Unterlassungsansprüche gegen Unternehmen geltend zu machen, die die Anforderungen nicht erfüllen.
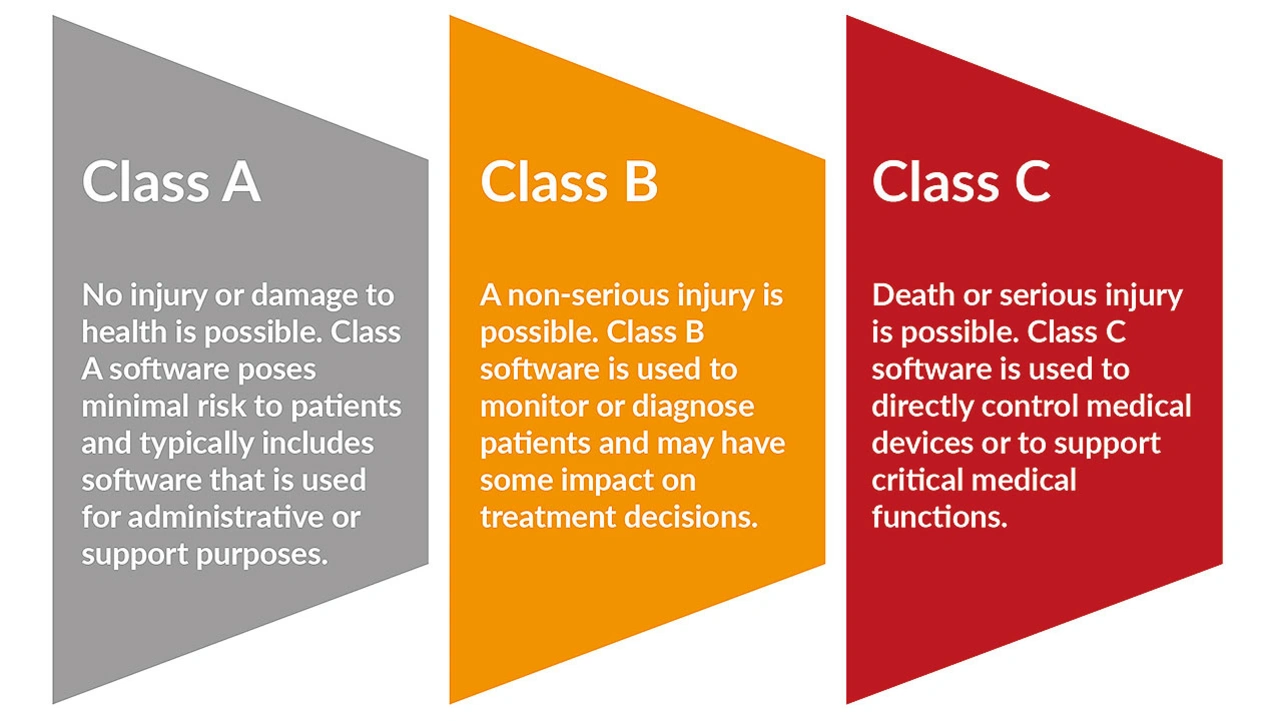
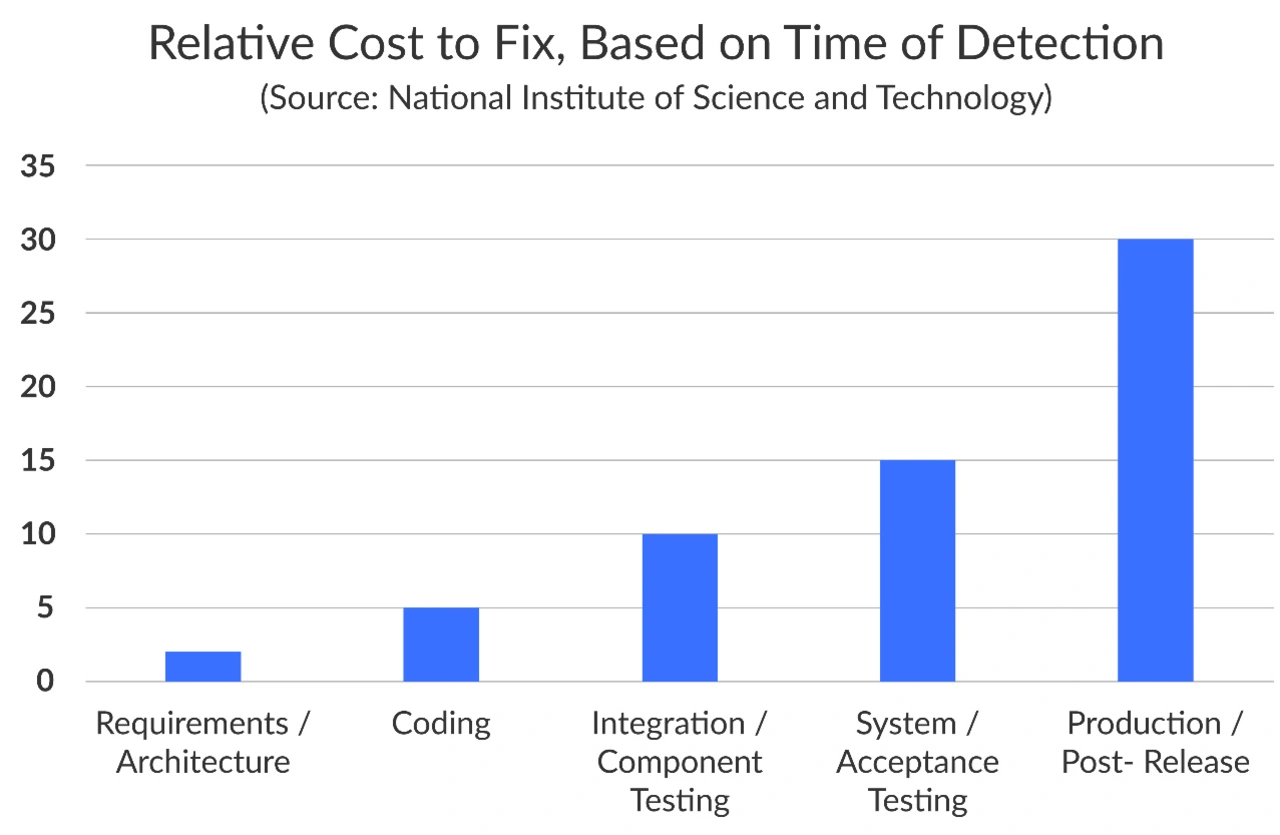
Um die Sicherheit, Wirksamkeit und Qualität ihrer Produkte zu gewährleisten, müssen Hersteller von Medizinprodukten je nach Sicherheitsklasse ihrer Geräte viele Anforderungen erfüllen (Bild 1). Im Folgenden werden die wichtigsten Aspekte der Einhaltung der FDA-Vorschriften erläutert, wobei der Schwerpunkt auf der Berücksichtigung von Sicherheitsbedenken bei Medizinprodukten und der Entwicklung von Kontrollen und Validierungsprotokollen für Hersteller liegt.
Sicherheitsfragen bei Medizinprodukten
Um die Anforderungen der FDA zu erfüllen und die Sicherheit der Patienten zu gewährleisten, müssen Hersteller von Medizinprodukten Sicherheitsbedenken effektiv angehen. Hier folgen Tipps zum Umgang mit Sicherheitsbedenken bei Medizinprodukten:
- Standardisierung des Codes und Durchführung einer Risikobewertung: Der erste Schritt auf dem Weg zu einem sicheren Medizinprodukt besteht darin, die Software nach standardisierten Programmierregeln zu schreiben. Bei bereits vorhandener Software kann der Code überprüft und bereinigt werden, um sicherzustellen, dass er akzeptablen Programmierstandards entspricht. Hersteller von Medizinprodukten können Lösungen zur statischen Codeanalyse einsetzen, um den Quellcode ihrer Softwareprogramme zu testen und zu analysieren. Neben der statischen Codeanalyse gibt es weitere Arten von Softwaretests, die dazu beitragen können, qualitativ hochwertige Software zu liefern, wie z. B. Integrationstests, Sicherheitstests, Unit-Tests, Systemtests und Usability-Tests. Die Bereinigung der Codebasis und die Durch- führung von Risikoanalysen helfen den Herstellern, potenzielle Sicherheitsschwachstellen in ihren Geräten zu erkennen. Dazu gehört auch die Bewertung von Risiken im Zusammenhang mit Datenschutzverletzungen, unberechtigtem Zugriff und der Integrität von Gerätefunktionen.
- Software-Updates und Patch-Management. Regelmäßige Updates und Patches der Gerätesoftware zur Behebung bekannter Schwachstellen. Rechtzeitige Aktualisierungen helfen, Sicherheitslücken zu vermeiden, die durch veraltete Software ausgenutzt werden könnten.
- Sichere Datenspeicherung um sicherzustellen, dass Patientendaten sowohl auf dem Gerät als auch während der Datenübertragung sicher gespeichert werden. Man sollte sichere Datenspeicherungsverfahren und branchenübliche Verschlüsselungsprotokolle verwenden, um die Privatsphäre der Patienten zu schützen und die Datenschutzbestimmungen einzuhalten.
- Zusammenarbeit mit Experten für Cybersicherheit und mit Fachleuten, die sich auf die Sicherheit von Medizinprodukten spezialisiert haben. Ihr Fachwissen kann den Herstellern helfen, potenzielle Sicherheitsrisiken zu erkennen und zu mindern.
___________________________________________________________________
Test-Driven Development (TDD) bei Smiths Medical
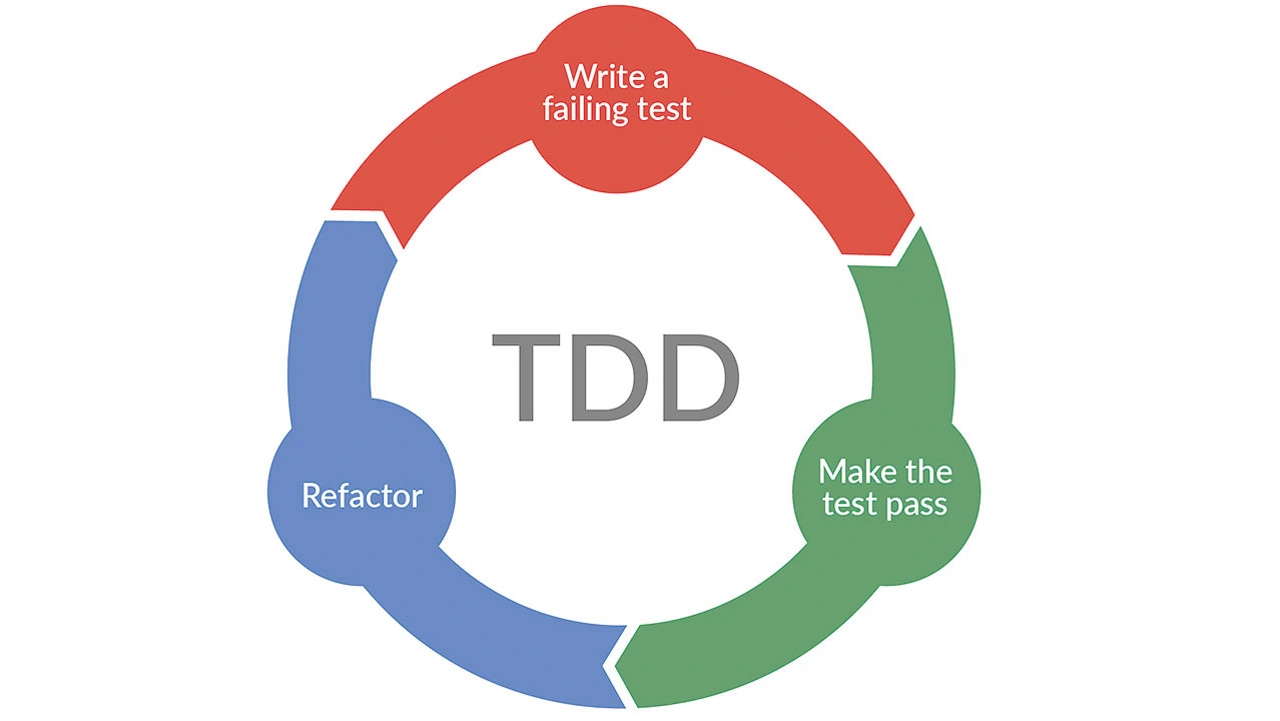
Smiths Medical, ein bekannter Hersteller von Infusionsgeräten, der heute zu ICU Medical gehört, setzt bei seiner Teststrategie stark auf automatisierte Tests. Frühere Versuche, umfassende Testwerkzeuge zu integrieren, waren nicht erfolgreich. Das Entwicklungsteam wollte aber den gesamten Testprozess verbessern. Es konzentrierte sich daher auf das sogeannten Unit Testing und Test Driven Development (TDD), eine Methodik, die Design, Test und Codeentwicklung integriert (Bild 3).
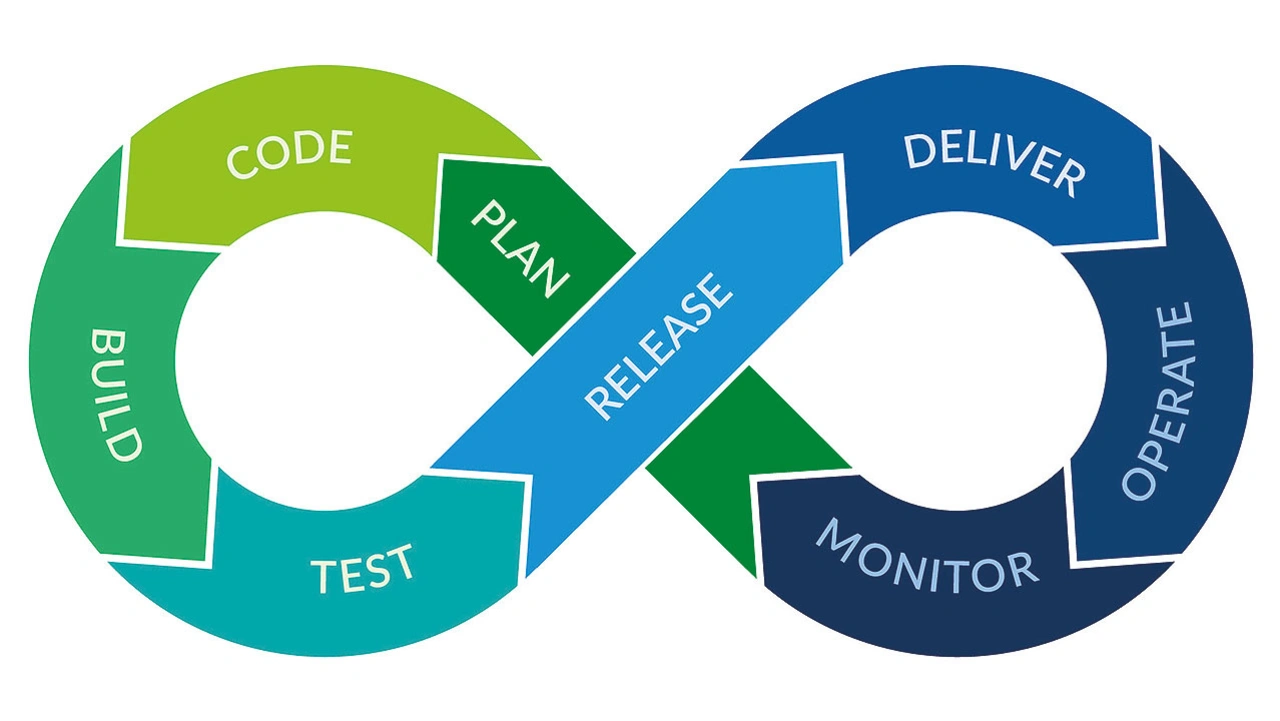
Smiths Medical benötigte ein Werkzeug, das nahtlos in die Testpipeline passt und die gesamte Entwicklungskultur verbessert. Mit Parasoft C/C++test konnte das Softwareteam nicht nur erfolgreich eine TDD-Implementierung durchführen, sondern profitierte auch von einer verbesserten Teststabilität, einer höheren Codeabdeckung und einem rationalisierten Tool-Qualifizierungsprozess, was insbesondere für sicherheitskritische Anwendungen von großer Bedeutung ist. Dadurch hat Smiths Medical seine Entwicklungsprozesse umgestaltet und das Testen zu einem integralen Bestandteil der Softwarepipeline gemacht (Bild 4). Dies stellt sicher, dass sichere und qualitativ hochwertige medizinische Geräte geliefert werden. _____________________________________________________________________
Designkontrollen und Validierungsprotokolle
Sorgfältige Designkontrollen und Validierungsprotokolle sind wichtige Komponenten, um sicherzustellen, dass Medizinprodukte die Anforderungen der FDA erfüllen. Diese Kontrollen unterstützen die Hersteller bei der Entwicklung sicherer und wirksamer Produkte, die den Patienten den beabsichtigten Nutzen bringen.
Nachfolgend sind die wichtigsten Überlegungen zur Implementierung von Designkontrollen und Validierungsprotokollen aufgeführt.
- Planung des Designs und der Entwicklung: Hersteller sollten einen klaren Design- und Entwicklungsplan erstellen, der den Umfang, die Ziele und die Erwartungen an das Medizinprodukt festlegt. Dieser Plan sollte auch das Risikomanagement, Studien zur Benutzerfreundlichkeit und die Einhaltung gesetzlicher Vorschriften umfassen.
- Designeingaben und -Ausgaben: Definition der Designeingaben, die die Anforderungen an das Produkt spezifizieren, und der Designausgaben, die die Eigenschaften des Produkts beschreiben. Es ist sicherzustellen, dass die Designinputs den Anforderungen der Benutzer und den gesetzlichen Anforderungen entsprechen.
- Verifizierung und Validierung: Anwendung strenger Verifizierungs- und Validierungsverfahren, um zu bestätigen, dass das Produkt die Konstruktionsanforderungen erfüllt und für die vorgesehene Verwendung geeignet ist. Dies beinhaltet die Prüfung und Bewertung der Leistung und Sicherheit des Produkts.
- Entwicklungsdokumentation (Design History File, DHF): Erstellen eines umfassenden DHF, in dem alle Konstruktions- und Entwicklungsaktivitäten aufgezeichnet sind, einschließlich der Dokumentation von Konstruktionskontrollen, Prüfergebnissen und Konstruktionsänderungen.
- Einreichung von Zulassungsanträgen: Die Einreichung korrekter und vollständiger Premarket Notifications (510(k)), De-Novo-Anträge oder Premarket Approval Applications (PMA) ist eine wichtige Voraussetzung für die Freigabe oder Zulassung von Medizinprodukten durch die FDA. Diese Anträge liefern der FDA die notwendigen Informationen über die Sicherheit und Wirksamkeit des Produkts und helfen ihr bei der Beurteilung der Verkehrsfähigkeit. Werden diese Anträge nicht korrekt ausgefüllt, kann dies zu Verzögerungen oder sogar zur Ablehnung des Produkts führen.
- Überwachung nach dem Inverkehrbringen (Post Market Surveillance, PMS). Die Einrichtung eines PMS-Systems ist entscheidend für die Überwachung und Meldung von unerwünschten Ereignissen, Beschwerden und Produktfehlfunktionen. Das Einhalten der PMS-Anforderungen hilft bei der Identifizierung von Sicherheitsproblemen und der raschen Umsetzung von Korrekturmaßnahmen. Es stellt sicher, dass die Hersteller die Leistung ihrer Produkte aktiv überwachen und sich mit allen Problemen befassen, die sich aus der Verwendung der Produkte ergeben.
Tools und Methoden zur Erfüllung der FDA-Anforderungen
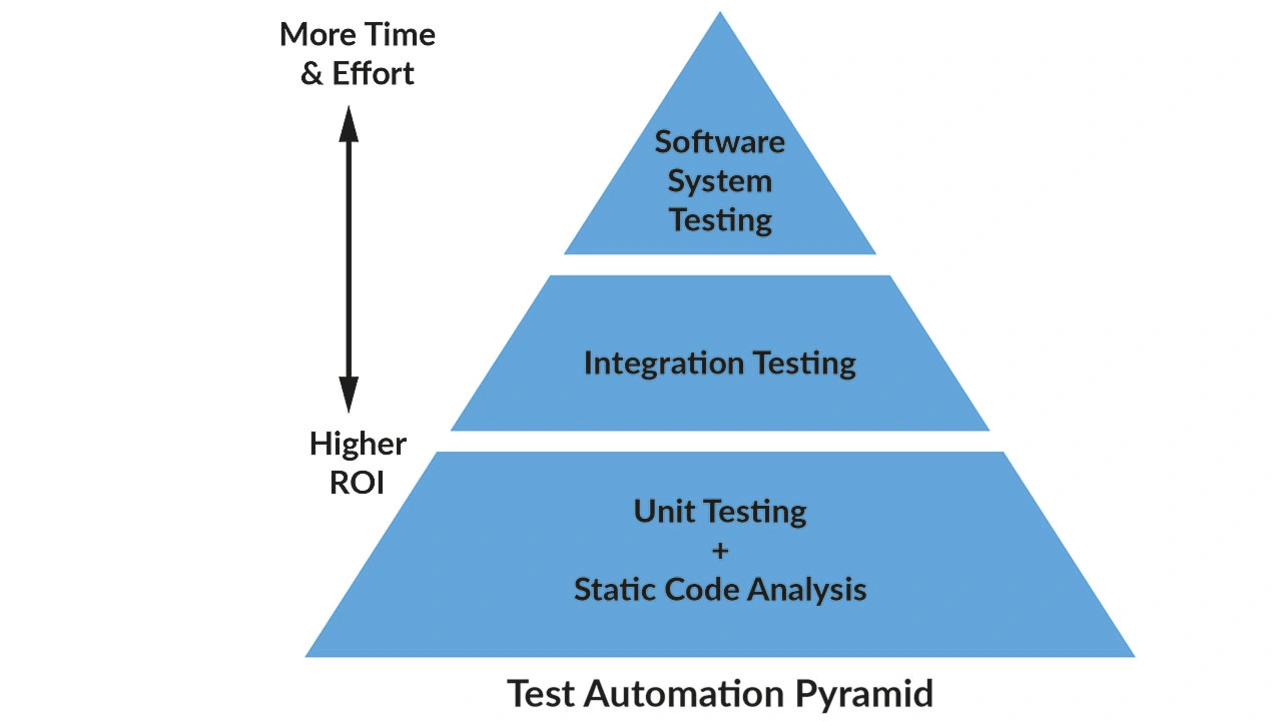
Hersteller können verschiedene Werkzeuge und Methoden einsetzen, um die Konformität mit den FDA-Vorschriften für Medizinprodukte zu erreichen und sicherzustellen (Bild 2). Folgend werden die wichtigsten vorgestellt, die bei der Erfüllung der FDA-Anforderungen helfen können.
Statische Analyse zur FDA-Konformität
Wenn es darum geht, Herstellern von Medizinprodukten zu helfen, die strengen Anforderungen der FDA zu erfüllen, spielt die statische Analyse eine entscheidende Rolle.
Hier sind fünf Gründe, warum die statische Analyse eingesetzt werden sollte, um die FDA-Anforderungen zu erfüllen.
- Früherkennung von Problemen: Statische Analysewerkzeuge können potenzielle Probleme wie Programmierfehler, Sicherheitsschwachstellen und Designfehler in einem frühen Stadium des Entwicklungsprozesses erkennen. Dies ermöglicht rechtzeitige Korrekturen und senkt das Risiko kostspieliger und zeitaufwändiger Änderungen zu einem späteren Zeitpunkt im Entwicklungszyklus. Empfohlene Programmierstandards für C und C++ sind MISRA C/C++ und CERT C/C++.
- Bessere Software-Qualität: Hochwertige Software ist für die Sicherheit und Wirksamkeit von Medizinprodukten von entscheidender Bedeutung. Die statische Analyse hilft den Entwicklern, sauberen, zuverlässigen und sicheren Code zu schreiben. Dies erleichtert nicht nur die FDA-Konformität, sondern verringert auch das Risiko von Problemen nach der Markteinführung und von Sicherheitsbedenken.
- Einhalten der Cybersecurity: Angesichts der zunehmenden Bedeutung der Cybersicherheit für Medizinprodukte legt die FDA großen Wert darauf. Statische Analysetools können Sicherheitslücken und Schwachstellen in der Software aufdecken und so die Einhaltung der FDA-Richtlinien zur Cybersicherheit gewährleisten.
- Vorschriftsmäßige Dokumentation: Die statische Analyse kann eine umfassende Dokumentation und Berichte liefern, die die gründliche Prüfung der Software und die Maßnahmen zur Behebung der gefundenen Probleme belegen. Diese Dokumentation ist für die Einreichung von Anträgen bei der FDA und für Inspektionen unerlässlich.
- Rationalisierte Validierung: Durch die frühzeitige Erkennung und Behebung potenzieller Probleme kann die statische Analyse den Validierungsprozess beschleunigen, der ein wichtiger Schritt zur Erfüllung der FDA-Anforderungen ist. Dies trägt dazu bei, Verzögerungen bei der Markteinführung von Medizinprodukten zu reduzieren.
Angemessenes Risikomanagement
Angesichts der Bedeutung des Risikomanagements für das Erreichen der Gesamtziele bzw. der FDA-Anforderungen ist es wichtig, sich damit zu befassen, wie ein angemessenes Risikomanagement erreicht werden kann.
- Risikobewertung: Die Risikobewertung bei der Herstellung von Medizinprodukten ist der erste Schritt zur Identifizierung und zum Verständnis der potenziellen Gefahren, die mit einem Medizinprodukt verbunden sind. Ihr Zweck ist die systematische Bewertung des Designs, der Zweckbestimmung, der Biokompatibilität und anderer Risikofaktoren des Produkts, um potenzielle Risiken zu ermitteln. In der Regel erfordert die Risikobewertung die Bildung eines funktionsübergreifenden Teams mit unterschiedlichen Fachkenntnissen, einschließlich Ingenieuren, klinischem Personal und Regulierungsexperten.
- Risikoanalyse: Sie zielt darauf ab, die identifizierten Risiken zu bewerten und zu priorisieren, indem ihnen eine Risikobewertung auf der Grundlage der Eintrittswahrscheinlichkeit und der Schwere des potenziellen Schadens zugewiesen wird. Dazu wird jedes Risiko durch die Bewertung seiner Eintrittswahrscheinlichkeit und seiner potenziellen Auswirkungen quantifiziert. Je nach Unternehmen kann eine Risikomatrix oder ein anderes Punktesystem zum Einsatz kommen, um die Risiken zu priorisieren. Punkte mit hohem Risiko werden als vorrangig für weitere Maßnahmen angesehen.
- Risikominderung: Dies beinhaltet die Entwicklung von Strategien, um identifizierte Risiken auf ein akzeptables Niveau zu reduzieren oder zu kontrollieren. Sie zielt darauf ab, die Sicherheit und Wirksamkeit des Medizinprodukts zu erhöhen. Risikominimierungsstrategien können verschiedene Formen annehmen, z. B. Konstruktionsänderungen, Sicherheitsmerkmale, verbesserte Materialien, verstärkte Anwenderschulung oder verbesserte Gebrauchsanweisungen. Ziel ist es, die identifizierten Risiken so zu behandeln, dass ihre Auswirkungen minimiert werden.
- Dokumentation: Eine angemessene Dokumentation ist für die Transparenz, die Rechenschaftspflicht und die Einhaltung der Vorschriften unerlässlich. Sie dokumentiert den Risikomanagementprozess und hilft beim Nachweis der gebotenen Sorgfalt gegenüber den Aufsichtsbehörden. Alle Aspekte des Risikomanagementprozesses, einschließlich Risikobewertungen, Analysen und Risikominderungspläne, sollten sorgfältig dokumentiert und während des gesamten Lebenszyklus des Produkts aufbewahrt werden.
- Kontinuierliche Verbesserung: Der Risikomanagementprozess sollte ein dynamisches und sich entwickelndes System sein. Kontinuierliche Verbesserungen stellen sicher, dass die Risikomanagementverfahren wirksam bleiben, wenn neue Risiken auftreten und sich die Marktbedingungen ändern. Regelmäßige Überprüfungen und Aktualisierungen ermöglichen es, Erfahrungen aus vergangenen Projekten einzubeziehen und an sich entwickelnde Technologien und Vorschriften anzupassen. Dies kann die Überprüfung und Überarbeitung von Risikobewertungen, Risikoanalysen und Risikominderungsplänen umfassen.
Statische Analyse für C/C++ beim Risikomanagement
Medizintechnik-Hersteller können die statische Analyse für C/C++ erfolgreich einführen, indem sie die folgenden Schritte befolgen:
1. Überprüfen der bestehenden Richtlinien im Unternehmen. Auch wenn diese manuell durchgesetzt werden müssen, sollten sie so weit wie möglich auf die mit dem statischen Analysetool bereitgestellten Checkern abgebildet werden. Ein ausgereiftes statisches Analysewerkzeug wird wahrscheinlich die meisten dieser Richtlinien abdecken. Für die verbleibenden Richtlinien, deren direkte Abbildung durch statische Analysetools nicht möglich ist, kann die Erstellung von benutzerdefinierten Checkern in Betracht gezogen werden.
2. Überprüfen der gängigen Programmierstandards, insbesondere derjenigen, die mit Blick auf die Sicherheit entwickelt wurden. Auswahl einer Untergruppe von Richtlinien, die das Team befolgen soll, wobei es ratsam ist, der Kategorisierung der Standards zu folgen und die Richtlinien auszuwählen, die als am wichtigsten erachtet werden. Zum Beispiel sollte man für CERT mit den L1 Richtlinien beginnen, und für MISRA C 2023 die obligatorischen Richtlinien prüfen.
3. Definieren der Konfiguration des statischen Analysewerkzeugs unter Berücksichtigung der unternehmensspezifischen Richtlinien und der ausgewählten Richtlinien wie z.B. CERT. Dabei nicht alle Checker auf einmal aktivieren, sondern mit einer kleinen Teilmenge beginnen, um die Entwickler nicht mit Verletzungen zu überfluten.
4. Sicherstellen, dass die Entwickler ihren Code sofort nach der Erstellung prüfen können. Sinnvoll ist auch, die statische Analyse in den CI/CD-Prozess zu integrieren.
5. Aktivieren weiterer Checker aus der Liste während des Entwicklungsprozesses und der Bereinigung des Quellcodes. Schließlich möchte man nachweisen können, dass man sich an alle ausgewählten Richtlinien gehalten hat und nicht auf halbem Wege stehen geblieben ist. Um den Überblick über den Prozess zu behalten und den aktuellen Fortschritt zu verstehen, ist ein zentrales Berichtssystem hilfreich, das die Testdaten zusammenfasst und die Arbeit der Entwickler überwacht. Ein Beispiel ist unten dargestellt:
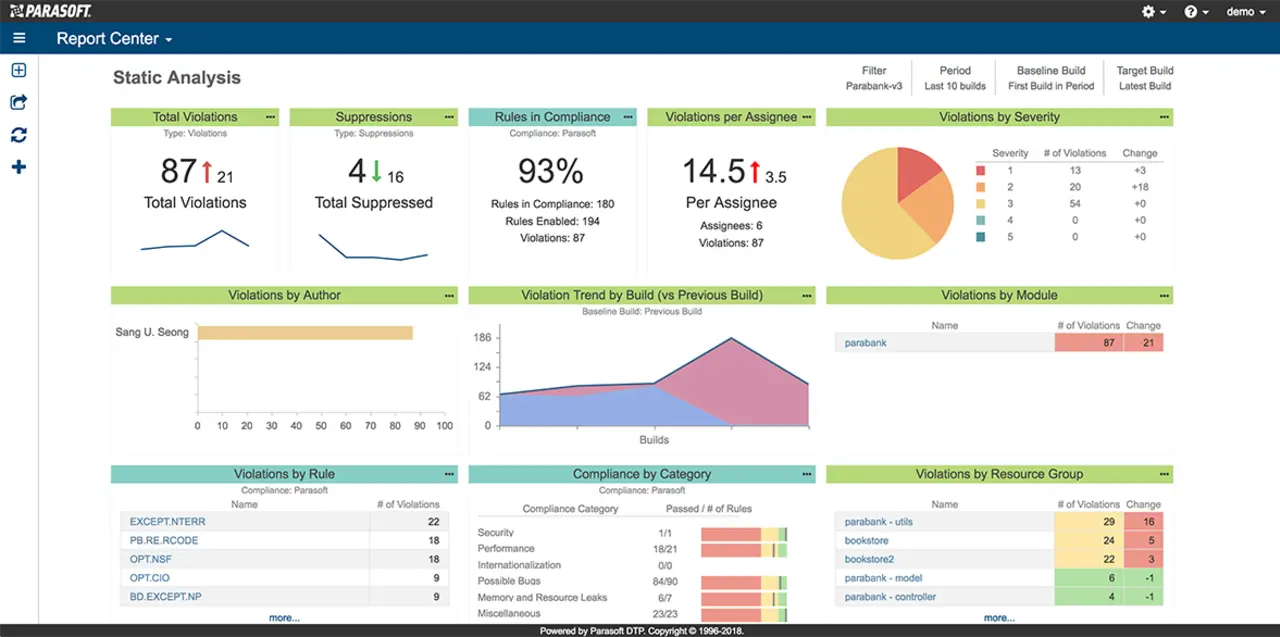
Da die Berichte über die statische Analyse Teil des Qualitätsmanagementsystems sind, kann man nicht irgendein Tool verwenden. Sondern die FDA verlangt, dass alle Werkzeuge, die für die Entwicklung und Verifikation von Software verwendet werden, für den beabsichtigten Zweck validiert sind. Es gibt verschiedene Möglichkeiten, die Eignung eines Tools für den Einsatz in der sicherheitskritischen Entwicklung nachzuweisen. Abhängig vom Risiko des Tools kann dies so einfach sein wie die Wiederverwendung eines Konformitätszertifikats oder der Abschluss des langwierigeren Prozesses der Werkzeugqualifizierung.
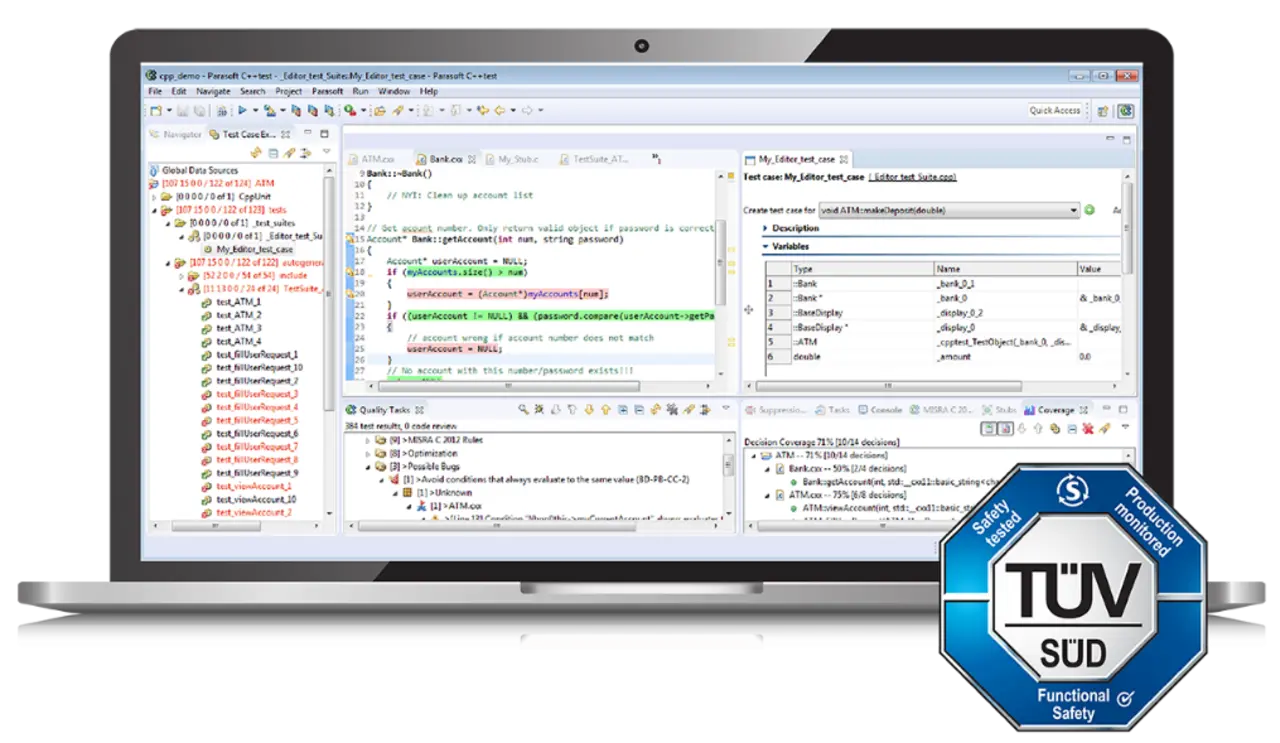
TÜV Süd-Zertifizierung für Medizin-Software
Für den Endanwender ist es am bequemsten, die Arbeit des Tool-Anbieters anzuerkennen und die Zertifizierung wiederzuverwenden, die für das Testwerkzeug von einer externen Zertifizierungsorganisation wie TÜV SÜD ausgestellt wurde. Beispielsweise verfügt Parasoft C/C++test über eine TÜV SÜD-Zertifizierung, die übernommen werden kann, um die Eignung für die Entwicklung von Software nach medizinischen Standards wie IEC 62304 nachzuweisen.
Tool-Qualifizierung durchführen
Bei Hochrisikogeräten, wie z.B. Klasse C Geräten, kann es notwendig sein, das Tool intern in der eigenen Entwicklungsumgebung zu validieren. Das Ziel besteht darin, nachzuweisen, dass das Tool in Übereinstimmung mit den betrieblichen Anforderungen arbeitet, die in der Entwicklungsumgebung des Projekts gesammelt wurden. Dies ist ein sehr mühsamer und zeitaufwendiger Prozess.
Am besten ist es, wenn der Tool-Anbieter diesen Prozess unterstützt und ein spezielles Tool-Qualifizierungs-Kit zur Verfügung stellt, das gut konzipierte Testfälle und das Automatisierungs-Framework enthält, um diese in der Entwicklungsumgebung des Projekts auszuführen und automatisch die Dokumentation zu erstellen, die als Nachweis für die Tool-Validierung dienen kann. Auch hier bietet Parasofts Vorzeigeprodukt C/C++test ein automatisiertes Tool-Qualifizierungs-Kit.
Anwendungsbeispiele und reale Anwendungen
Es gibt zahlreiche Case Studies über reale Anwendungen der statischen Analysetools von Parasoft. Folgende Fallstudien veranschaulichen, wie die Softwaretestlösungen von Parasoft den Herstellern medizinischer Geräte geholfen haben, ihre spezifischen Herausforderungen zu bewältigen und ihre Ziele zu erreichen. Sowohl Inovytec als auch Smiths Medical konnten durch die Zusammenarbeit mit Parasoft bemerkenswerte Verbesserungen in Bezug auf Codequalität, Konformität und Testeffizienz erzielen.
Inovytecs Weg zur FDA 510(k)-Zertifizierung |
|---|
Inovytec, ein Unternehmen, das sich auf die Herstellung medizinischer Geräte spezialisiert hat, musste die FDA 510(k)-Zertifizierung für das Beatmungsgerät Ventway Sparrow erhalten. Die Herausforderung bestand darin, einen sauberen Code zu liefern und gleichzeitig die FDA-Anforderungen zu erfüllen. Das Softwareentwicklungsteam von Inovytec passte Parasoft C/C++test an die strengen Anforderungen der FDA an. Jedes Mal, wenn eine neue Version der Software veröffentlicht werden sollte, wurde sichergestellt, dass die statische Analyse von Parasoft so konfiguriert war, dass sie den FDA-Anforderungen entsprach. Das Ergebnis war nicht nur eine verbesserte Codequalität, sondern auch ein durchschlagender Erfolg bei der 100%igen Erfüllung der FDA 510(k)-Zertifizierungsregeln und -richtlinien. Parasoft hat sich bei Inovytec als bevorzugte Testlösung durchgesetzt und die Zusammenarbeit mit ESL, einem Vertriebspartner für Parasoft-Produkte in Israel, bot bei Bedarf wichtige Unterstützung und Fachwissen. Wie MedTech-Firmen die Herausforderungen bei der Softwareentwicklung für Medizinprodukte mit einem integrierten SDLC-Ansatz meistern können, zeigt ein Whitepaper von Parasoft. |
Herausforderungen der FDA-Validierung lösen
Der FDA-Validierungsprozess kann für Hersteller von Medizinprodukten komplex und schwierig sein. Es gibt jedoch eine Reihe von Schritten, die Hersteller durchführen können, um diese Herausforderungen zu meistern.
- Frühzeitig beginnen - Der FDA-Validierungsprozess sollte früh im Produktentwicklungszyklus beginnen. Dadurch haben die Hersteller mehr Zeit für die Planung und Durchführung des Validierungsprozesses und können potenzielle Probleme frühzeitig erkennen und angehen.
- Einen risikobasierten Ansatz verwenden - Eine Risikobewertung des Produkts sollte die Grundlage für den Validierungsprozess der FDA bilden. Hersteller sollten ihre Validierungsbemühungen auf die kritischsten Komponenten und Funktionen des Produkts konzentrieren.
- Einsatz von qualifiziertem Personal - Nur qualifiziertes Personal, das Erfahrung mit den FDA-Vorschriften und bewährten Validierungsverfahren aufweist, sollte den FDA-Validierungsprozess durchführen.
- Dokumentation des Validierungsprozesses - Hersteller sollten den FDA-Validierungsprozess sorgfältig dokumentieren. Auf diese Weise können sie gegenüber der FDA nachweisen, dass sie ihr Produkt in Übereinstimmung mit den FDA-Vorschriften validiert haben.
Nächste Schritte für die Validierung bei der FDA
Die FDA stellt Herstellern von Medizinprodukten wichtige Validierungsrichtlinien zur Verfügung, um die Produktqualität und Patientensicherheit zu gewährleisten. Da sich die Vorschriften und Richtlinien ständig weiterentwickeln, ist es wichtig, mit diesen Änderungen Schritt zu halten und die zukünftige Konformität zu planen.
Vorbereitung auf zukünftige FDA-Richtlinien
Weil die FDA ihre Richtlinien für Hersteller von Medizinprodukten laufend aktualisiert und überarbeitet, empfehlen sich folgende Maßnahmen für Hersteller von Medizinprodukten:
- Regelmäßiger Check der FDA-Website auf Aktualisierungen der für ihre Produkte relevanten Leitliniendokumente, die die FDA dort veröffentlicht.
- Teilnahme an Workshops und Webinaren der FDA, um über die neuesten Überlegungen der FDA zur Regulierung von Medizinprodukten auf dem Laufenden zu bleiben.
- Vernetzung mit anderen Herstellern von Medizinprodukten zum Austausch von Informationen und Best Practices im Hinblick auf die Einhaltung der FDA-Vorschriften.
Kontinuierliche Konformität sicherstellen
Hersteller von Medizinprodukten müssen die kontinuierliche Einhaltung der FDA-Vorschriften sicherstellen. Um dies zu erreichen, sollten Hersteller
- ein Qualitätssicherungssystem einführen und aufrechterhalten, das die Anforderungen der FDA-Verordnung über Qualitätssysteme erfüllt.
- regelmäßige Audits ihres Qualitätssystems durchführen, um alle Bereiche, in denen die Anforderungen nicht erfüllt werden, zu identifizieren und zu korrigieren.
- MDR-Berichte bei der FDA einreichen. Hersteller sind verpflichtet, der FDA Medical Device Reports (MDRs) über unerwünschte Ereignisse im Zusammenhang mit ihren Produkten vorzulegen. Durch die Übermittlung von MDR-Berichten an die FDA können Hersteller die FDA dabei unterstützen, Sicherheitsprobleme im Zusammenhang mit ihren Produkten zu erkennen und zu beheben.
Im Laufe der Zeit hat sich gezeigt, dass die Einhaltung der FDA-Vorschriften sehr streng und zeitaufwändig ist. Bei der Bewältigung dieser Herausforderungen bieten statische und dynamische Analysetools wertvolle Unterstützung. Mit den hochentwickelten Funktionen der statischen Analysewerkzeuge können Probleme in Software und Code bereits in einem frühen Stadium des Entwicklungsprozesses erkannt und behoben werden.
Die statische Analyse einzuführen, ist ein wichtiger Schritt, der Zeit kostet und die Entwickler belastet. Dennoch hat sie sich bewährt, um das System gegen bösartige Angriffe zu schützen. Wird die statische Analyse zusammen mit gut durchdachten Sicherheitsrichtlinien eingesetzt, können Systeme entwickelt werden, die auch unvorhergesehenen Angriffen standhalten. (uh)
