Der Gesetzgeber redet kräftig mit
Software geändert - was nun?
Fortsetzung des Artikels von Teil 1
Strafe muss sein
Verstößt ein Hersteller mit seinen Änderungen gegen die Auflagen der jeweiligen Gesundheitsbehörden, drohen unterschiedliche Strafen. Beispielsweise sind innerhalb Europas die Strafmaßnahmen für Verstöße gegen die »93/42/EWG über Medizinprodukte« in den nationalen Umsetzungen der Richtlinie verankert. Das deutsche Medizinproduktegesetz (MPG) listet Freiheitsstrafen bis zu drei Jahren oder eine Geldstrafe. Verletzungen der US-amerikanischen Vorschriften führen dagegen zu einem »Warning Letter«, der unverzüglich zur Ergreifung von Maßnahmen auffordert.
Zuwiderhandlungen gegen diese Aufforderung werden mit Freiheitsstrafe und/oder Bußgeld geahndet und ziehen für ausländische Hersteller einen »Import Alert« nach sich. Damit kann die FDA die Einführung eines Produkts in die USA verhindern, bis sie selbst die Änderungen am Produkt freigibt.
Nicht immer meldepflichtig
Aufgrund der unterschiedlichen Auswirkungen auf ein Medizinprodukt erfordern Änderungen nicht immer eine erneute Freigabe durch die Zulassungsbehörden. Hier ist zwischen meldepflichtigen und nicht meldepflichtigen Änderungen zu unterscheiden. Grundsätzlich gelten alle Modifikationen, die ein zugelassenes Medizinprodukt verändern und somit dessen Sicherheit und Leistungsfähigkeit berühren, als meldepflichtig.
Maßgeblich ist die Entscheidung, ob die Änderung »substanzieller« Natur ist. Die Veränderung der Farbe eines Buttons kann, muss aber nicht zwangsweise dazu führen, dass eine erneute Freigabe notwendig wird. Verändert beispielsweise die Farbe des Buttons die Benutzerschnittstelle einer Hauptbedienfunktion, ist zu entscheiden, ob diese Änderung die Sicherheit des Medizinprodukts beeinflusst.
Diese Entscheidung liegt jedoch allein beim Hersteller. Für die Entscheidung, welche Änderungen meldepflichtig sind, existieren je nach Zulassungsgebiet unterschiedliche Regelungen und Entscheidungshilfen. Innerhalb Europas unterstützen die Benannten Stellen durch eine Sammlung von Beispielen melde- und nicht meldepflichtiger Änderungen bei der Einteilung der Änderungen.
Die »European Association of Notified Bodies for Medical Devices« listet in ihrer Empfehlung »Reporting of design changes and changes of the quality system (NB-MED/2.5.2/Rec2)« die Punkte auf, die bei der Entscheidung über die Meldepflichtigkeit zu berücksichtigen sind. So gilt es beispielsweise zu überprüfen, ob die Änderung den bestimmungsgemäßen Gebrauch des Medizingerätes und die Nutzergruppe verändert oder ob neue beziehungsweise geänderte Risiken zu betrachten sind.
Den Herstellern wird empfohlen, einen Prozess zur Beurteilung und Dokumentation von Änderungen zu definieren und die Vorgehensweise im Falle einer Meldepflicht an die Benannte Stelle festzulegen. Zudem listet die Empfehlung Beispiele meldepflichtiger und nicht meldepflichtiger Änderungen auf und weist speziell für Software auf neue Funktionen und neue Algorithmen hin. In den USA stellt die FDA als zuständige Behörde Flussdiagramme als Entscheidungshilfe bereit, die in der Guideline »Deciding When to Submit a 510(k) for a Change to an Existing Device« beschrieben sind.
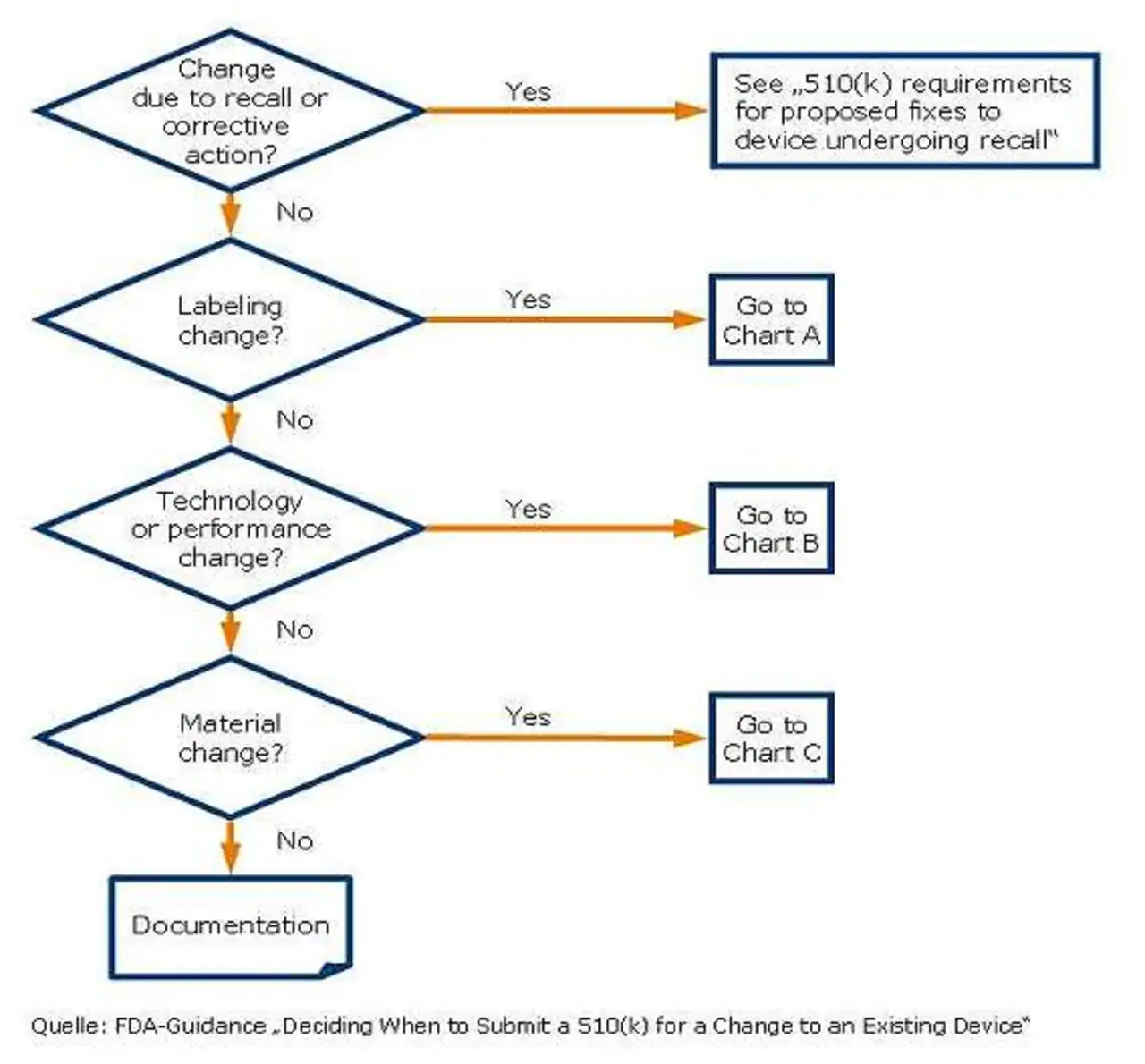
Bei Anwendung der Flussdiagramme werden die Änderungen in vier Klassen (labeling, technical, performance, material) unterteilt, für die jeweils eigene Flussdiagramme gelten (Bild 2). Die FDA empfiehlt, ein geändertes Produkt mit der letzten freigegebenen Version zu vergleichen. Es wird explizit darauf hingewiesen, dass auch die Summe vieler nichtmeldepflichtiger Änderungen in der Gesamtbetrachtung eine meldepflichtige Änderung darstellen kann.
Ebenso wird darauf Wert gelegt, dass jede Änderung separat zu betrachten ist. Unabhängig vom Ausgang der Entscheidung muss der Hersteller die Änderung in dem »Device Master Record« dokumentieren und die Anwendung der Flussdiagramme inklusive der abgeleiteten Ergebnisse festhalten. Kanada führt alle Arten meldepflichtiger Änderungen direkt im Gesetz unter Artikel 34 auf.
Dazu gehören neben Änderungen am Gerät selbst auch die Änderung des Geräte- und des Herstellernamens, ebenso wie Änderungen an Bezeichnungen, die zur Identifizierung eines Gerätes oder einzelner Gerätekomponenten dienen. Auch das japanische Gesundheitsministerium nutzt die Unterscheidung in meldepflichtige (»partial changes«) und nichtmeldepflichtige Änderungen (»minor changes«). Als Entscheidungshilfe sind Beispiele für meldepflichtige Änderungen im Anhang 2 der Guideline »Points to Consider When Applying for Marketing Approval for Medical Devices: PFSB/ELD/OMDE Notification No. 0216001« aufgeführt.
Für »minor changes« reicht eine schriftliche Mitteilung an die Behörden innerhalb von 30 Tagen nach Änderung.
Richtig Meldung machen
Stuft der Hersteller eine Änderung als meldepflichtig ein, muss er die für die jeweilige Zulassungsbehörde geltenden Vorgaben beachten. Auf dem europäischen Markt muss der Hersteller Änderungen am Produkt den zuständigen Benannten Stellen durch eine Änderungsmitteilung melden. Für die Änderungsmitteilung stellt beispielsweise der TÜV Süd ein eigenes Formblatt bereit.
Der Änderungsantrag muss eine Beschreibung der Änderung enthalten, den Grund der Änderung und die Auswirkungen auf die Ergebnisse der Risikoanalyse sowie der »Grundlegenden Anforderungen« laut Anhang II der »Medical Device Directive«. Wird die gemeldete Änderung akzeptiert, stellt die Benannte Stelle entweder ein neues Zertifikat oder eine Ergänzung zu einem bestehenden Zertifikat aus.
Hat ein Hersteller auf dem US-amerikanischen Markt entschieden, dass eine signifikante Änderung vorliegt, muss er eine neue »Premarket Notification 510 (k)« einreichen. Mittels eines »510 (k)« zeigen Hersteller, dass ihr geändertes Produkt einem bereits zugelassenen Produkt substanziell gleicht und somit genauso sicher und effektiv wie das bereits zugelassene Produkt ist. Es besteht die Möglichkeit, ein abgekürztes Verfahren zu durchlaufen, das speziell für Änderungen an Medizinprodukten geschaffen wurde.
Dieses »Special 510 (k)« genannte Verfahren ermöglicht die Freigabe seitens der FDA innerhalb von 30 Tagen. Voraussetzung hierfür ist jedoch, dass die Modifikation am Medizinprodukt weder die medizinische Zweck-bestimmung noch die technologischen Grundlagen verändert. Auf dem kanadischen Markt gibt das dortige Gesundheitsministerium Änderungen frei. Ein Hersteller muss hierzu bei Health Canada einen Änderungsantrag (Medical Device Licence Amendment) stellen.
Für jede Risikoklasse sowie für Änderungen am Produkt- oder Herstellernamen gibt es einen eigenen Antrag. Die einzureichenden Dokumente beschränken sich jedoch lediglich auf die für die Änderung relevanten Informationen. Meldepflichtigen Änderungen auf dem japanischen Markt benötigen Prüfung und Freigabe durch die PMDA. Ohne Freigabe durch die dem »Ministry of Health, Labour and Welfare« untergeordnete Behörde darf ein geändertes Produkt nicht auf dem japanischen Markt vertrieben werden. Welche Dokumente für ein »partial change« einzureichen sind, ist in Kapitel IV, Punkt 3 »Application for Partial Change of Approved Information« der Notification No. 0216001 gelistet.
Schwellenländer - Beispiel China
China, als Schwellenland auf dem Weg zur größten Volkswirtschaft der Welt, wird als Markt für Medizintechnikhersteller immer wichtiger. Vorschriften für die Zulassung von Medizinprodukten und das Vorgehen bei Änderungen finden sich in den »Regulations for the Supervision and Administration of Medical Devices« sowie in der Guideline »Initial registration of import products«. Die gesetzlich notwendige Dokumentation umfasst die Beschreibung der Änderung am Medizinprodukt und einen Vergleich mit dem Vorgängermodell.
Der Autor:

- Software geändert - was nun?
- Strafe muss sein
Lesen Sie mehr zum Thema
